







Two scientists whose work ushered in the first approved therapy using the gene-editing tool CRISPR have won the $3 million Breakthrough Prize in Life Sciences.
The prize winners — Dr. Swee Lay Thein, of the National Heart, Lung and Blood Institute (NHLBI), and Dr. Stuart H. Orkin, of Harvard University — shared the award for basic research that led to the development of a gene therapy that treats the blood disorders sickle cell disease and beta-thalassemia.
The Breakthrough Prize in Life Sciences has been awarded since 2013 to recognize accomplishments in the life sciences.
Deadly blood disorders
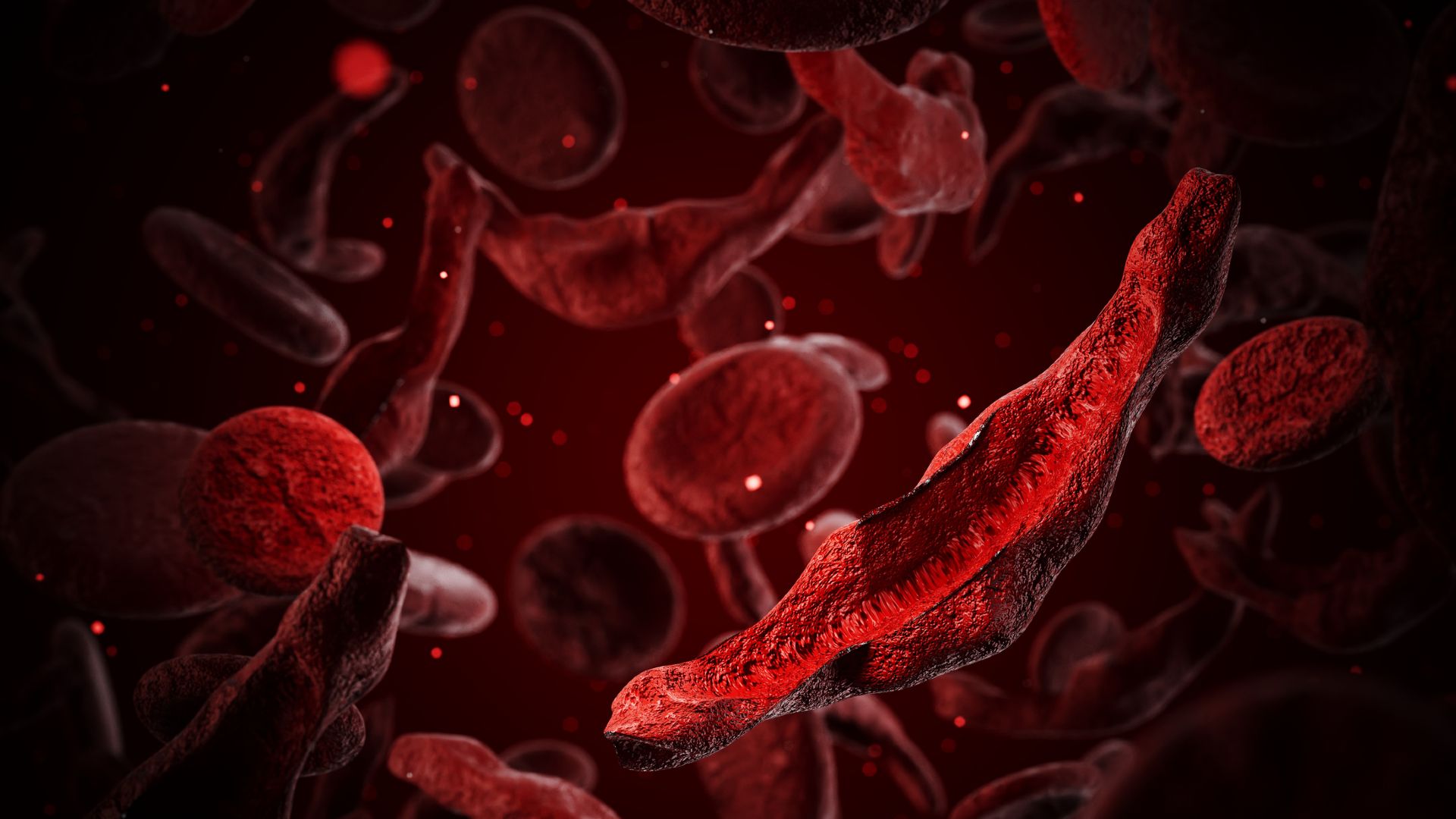
Sickle cell disease affects around 7 million to 8 million people globally, predominantly in Africa. In people with the disorder, red blood cells take on a characteristic crescent shape because hemoglobin, the oxygen-carrying molecule inside the cells, forms stiff, long fibrils that deform the cells. These sickled cells stick to one another, triggering blood clots, and they also burst and die easily, causing low red-blood-cell counts.
Patients often face excruciating episodes of pain, known as “crises,” when the red blood cells block blood vessels. These blockages can damage organs like the lungs, liver and spleen. The blockages in the lungs can also trigger “acute chest syndrome,” which depletes oxygen levels and is the leading cause of death in sickle cell patients.
In beta-thalassemia, the body either does not make — or makes lower amounts of — one portion of the hemoglobin molecule, meaning people with severe forms of the disease must receive blood transfusions for life. Casgevy is approved to treat this severe form of the disease.
Thein, who is a senior investigator at the NHLBI, began her work in the 1980s trying to figure out why some people with these disorders had much milder forms of the diseases than others.
The question had emerged decades earlier, when Dr. Janet Watson, a New York-based pediatrician, showed that infants who later developed sickle cell disease didn’t show symptoms and had red blood cells that did not sickle.
Once children were toddlers, symptoms of the disease emerged.
Follow-up work showed that people produce different types of hemoglobin at different stages of development: “Fetal hemoglobin” is produced in the womb, and its production is turned off as babies mature and “adult hemoglobin” takes over.


“I started collecting families — patients — with mild thalassemia, to try to at least unravel the genetics behind it,” Thein told Live Science. “It seemed obvious that they have an innate ability, or natural ability, to continue producing fetal hemoglobin.”
She analyzed the genes of several families that had a history of disease, including a family of Indian origin that included more than 200 members, spanned seven generations and lived on multiple continents.
Repressing the repressor
A crucial insight came from a study of pairs of identical and fraternal twins who made either very high or very low levels of fetal hemoglobin. This enabled Thein and her colleagues to identify gene variants that affected fetal hemoglobin production. They zeroed in on a region of a gene on chromosome 11 called BCL11A.
Thein’s team found that the gene turns off the production of fetal hemoglobin as babies grow. “It’s a repressor,” Thein said. But when people carried certain versions of BCL11A, the repressor didn’t repress and fetal hemoglobin production continued at high levels throughout life.
From there, it wasn’t a dramatic leap to conclude that repressing the repressor could be a good strategy to treat people with severe versions of sickle cell disease or beta-thalassemia. Orkin’s research proved pivotal in making that leap.
Orkin — who is a pediatric hematologist and oncologist at Boston Children’s Hospital, Dana-Farber Cancer Institute, Harvard Medical School, and Howard Hughes Medical Institute — showed how the repressor mediated the switch to adult hemoglobin, and that gene editing could target the region.
The biotech company Vertex then used the cut-and-paste gene-editing tool CRISPR to snip out the repressor region of BCL11A.
This work eventually led to the development of Casgevy. Administering the therapy involves extracting a person’s bone marrow cells, editing the BCL11A repressor using CRISPR, and then reinfusing the gene-edited bone marrow cells back into the patient. The edited cells begin to make red blood cells with high levels of fetal hemoglobin.


It’s the first “functional cure” for sickle cell disease, and it has transformed the lives of the few who have received it. But it’s not a cure available for everyone with the disease, and there are some drawbacks, Thein said. The treatment process itself can take up to a year, costs a few million dollars, and requires harsh chemotherapy to make space in the bone marrow for the gene-edited stem cells to take root.
“Physically, it’s very grueling for the patient,” Thein said.
In addition, sickle cell disease and beta-thalassemia predominantly affect people in Africa, Asia and the Mediterranean, where the resources and facilities needed for such treatment may not be available. As a result, scientists working on gene therapy are pivoting to an “in vivo” approach, which involves “actually injecting the gene editing machinery into the patient,” Thein said. This would cut out the need to extract, edit and reinfuse bone marrow cells.
Ultimately, the need for more drugs — including cheaper, more easily delivered pills, shots or infusions — is still pressing, Thein said.
Thein has studied a drug called Mitavipat. The drug, which is currently approved for the treatment of the blood disease pyruvate kinase deficiency and beta thalassemia, seems to work by improving the overall metabolic health of red blood cells, Thein said.
Some of the patients on this drug have “been on this treatment and with me for six years, and it has really made quite a big difference,” she said, but further tests are needed to approve its use in people with sickle cell disease.









